

Introduktion till von Willebrands sjukdom
Introduktion till von Willebrands sjukdom
Vad är von Willebrands sjukdom?
Von Willebrands sjukdom (VWS) är den vanligaste orsaken till medfödd ökad blödningsbenägenhet. Det finns flera olika typer av VWS. Sjukdomen orsakas av brist på eller nedsatt funktion hos ett protein (äggviteämne) som benämns ”von Willebrandfaktorn” (VWF), som tillverkas i blodkärlens väggar.
VWF finns i blodet och är nödvändig för normal blodstillning, s.k. koagulation. Det finns olika långa VWF-kedjor i blodet och dessa fungerar också som bärarprotein för koagulationsfaktor 8 (FVIII). Vid brist på VWF kan man därför också få en låg FVIII-nivå.
När det inte finns tillräckligt med VWF i blodet, eller om VWF inte fungerar normalt, tar det längre tid för blodet att koagulera (levra sig) och blödningar att upphöra.
Det finns också en förvärvad (dvs. inte medfödd eller ärftlig) form av VWS, som kan uppstå senare i livet. Denna typ av VWS är betydligt ovanligare än den ärftliga formen. Förmågan att bilda VWF är då normal, men av någon anledning inaktiveras VWF helt eller delvis.
Den här informationssidan handlar framför allt om den medfödda formen av VWS.
Hur koagulerar blodet under normala förhållanden?
Blodet cirkulerar i hela kroppen i ett utbrett nätverk av blodkärl. Om någon av kroppens vävnader skadas kan blodkärlen gå sönder och blod läcka ut. Då man skär sig på en kniv blir blödningen synlig för ögat. Om blödningen ligger under hudytan kan den synas som ett blåmärke. Djupare skador kan orsaka inre blödningar som inte syns.
Kroppens koagulationssystem får blodet att levra sig och bilda ett koagel som stoppar blödningen. Koagulationsprocessen kan indelas i tre steg:
Koagulationsprocessen
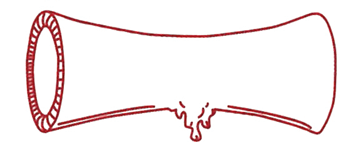 |
Steg 1 Blodkärlen dras samman (kontraheras) för att minska blodflödet till det skadade området. |
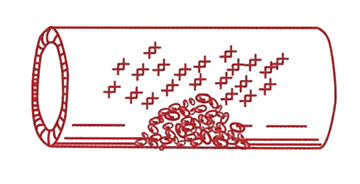 |
Steg 2 Blodplättar (trombocyter) är små blodkroppar med en diameter mindre än 1/10.000-dels centimeter som cirkulerar i blodet. Blodplättar fastnar och breder ut sig över skadan i kärlväggen. VWF fungerar som ett klister som håller blodplättarna på plats vid kärlskadan. Blodplättarna sammanhållna av VWF bildar en trombocytplugg som stoppar blödningen tillfälligt. |
 |
Steg 3 För att få en varaktig blodstillning krävs att blodet dessutom koagulerar (levrar sig), så att ett fast koagel bildas. Detta sker genom att olika koagulationsfaktorer reagerar med varandra på ytan av trombocytpluggen. Därigenom bildas ett nätverk bestående av långa fibrintrådar. Fibrinnätet knyter samman trombocytpluggen till ett fast koagel som varaktigt tätar läckan i blodkärlsväggen.
|
 |
Normal von Willebrandfaktor, som har olika långa kedjor. |
Hur påverkar von Willebrands sjukdom den normala koagulationen?
VWF påverkar flera steg i koagulationsprocessen:
Koagulationsprocessen
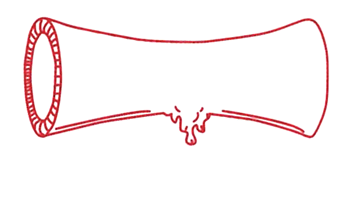 |
Steg 1 Blodkärlen kontraheras normalt. |
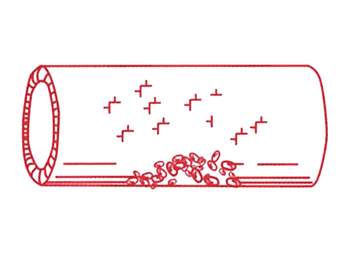 |
Steg 2 En person med VWS har för lite VWF eller nedsatt funktion hos VWF. Det gör att blodplättarna får svårare att bilda en trombocytplugg. Trombocytpluggen blir liten och skör. |
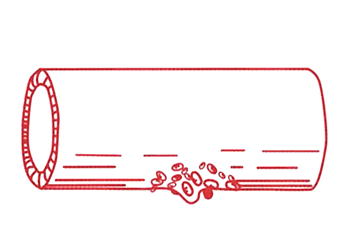 |
Steg 3 Fibrinnätet bildas långsammare och blir skörare än normalt, dels därför att trombocytpluggen är sämre, dels p.g.a. att personer med VWS även kan ha brist på faktor VIII (FVIII), som är en av de s.k. koagulationsfaktorerna. FVIII är mycket skör och inaktiveras lätt, men skyddas i normala fall av VWF, eftersom VWF är ett bärarprotein för FVIII. Vid brist på VWF blir det därför även brist på FVIII. |
 |
Onormal von Willebrandfaktor |
Hur vanlig är von Willebrands sjukdom?
Vissa forskare tror att ärftlig form av VWS kan finnas hos så många som 1 av 100 individer i normalbefolkningen. Eftersom många har mycket milda besvär har sjukdomen endast diagnostiserats hos ett fåtal.
Vem kan ha von Willebrands sjukdom?
Både kvinnor och män kan ha VWS. Kvinnor märker dock tydligare att de har sjukdomen, eftersom VWS kan ge upphov till rikliga menstruationsblödningar och onormalt stora blödningar efter förlossningar. Eftersom VWS är en medfödd och ärftlig sjukdom finns sjukdomen redan från födseln. Därför kan även barn ha VWS.
Kan von Willebrands sjukdom överföras från föräldrar till barn?
Ja. Om en eller båda föräldrarna har VWS kan sjukdomen ärvas av barnen. Ärftlighet kan du läsa mer om här.
Varför heter sjukdomen von Willebrands sjukdom (och lite historia)?
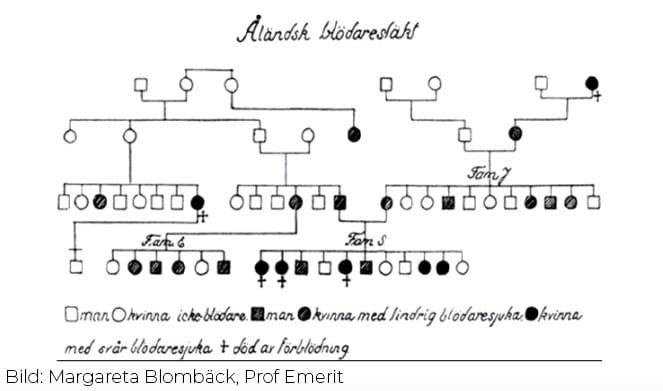
 VWS har uppkallats efter en finsk läkare, Erik von Willebrand (bilden till vänster), som beskrev tillståndet 1926 hos en familj på Åland. Han insåg att VWS inte är samma sjukdom som klassisk blödarsjuka (hemofili), som är en annan ärftlig blödningsbenägenhet, vilken huvudsakligen drabbar män. Redan 1852 beskrevs liknande familjer i USA och Europa med såväl kvinnliga som manliga blödare, men sjukdomen klassades då som hemofili.
VWS har uppkallats efter en finsk läkare, Erik von Willebrand (bilden till vänster), som beskrev tillståndet 1926 hos en familj på Åland. Han insåg att VWS inte är samma sjukdom som klassisk blödarsjuka (hemofili), som är en annan ärftlig blödningsbenägenhet, vilken huvudsakligen drabbar män. Redan 1852 beskrevs liknande familjer i USA och Europa med såväl kvinnliga som manliga blödare, men sjukdomen klassades då som hemofili.
Erik von Willebrand kallade själv tillståndet för ”hereditär pseudohemofili” eller ”konstitutionell trombopati”. I artikeln från 1926 visas en släkttavla med 57 individer från två relaterade familjer över fyra generationer. Av de elva barnen i familjen S var det endast fyra som inte hade blödningssymtom. Ärftlighetsanalysen som presenterades i artikeln föreslog att det handlade om dominant ärftlighet, som du kan läsa mer om här.
Erik von Willebrand och dennes forskargärning presenteras av de finska läkarna R. Lassila och O. Lindberg i en artikel i Haemophilia (2013), 19, 643647.
Hur allvarlig är von Willebrands sjukdom?
Det beror på typ och svårighetsgrad av sjukdomen. De flesta har VWS i mycket mild form och märker knappt att de har sjukdomen förrän de får en kraftig blödning i samband med en olycka eller en operation.
Vissa personer med VWS blöder ganska ofta. Det rör sig då oftast om näsblödningar eller rikliga menstruationsblödningar. Personer med mild VWS får svåra blödningar endast om de skadas eller opereras utan förbehandling. Personer med svår form av VWS kan däremot få svåra blödningar utan att de råkat ut för en olycka. Det kan röra sig om blödningar i leder (t.ex. knäleder) eller svåra mag- eller tarmblödningar.
Svår form av VWS är mycket ovanlig. Endast ett 50-tal fall med den svåraste formen (typ 3) har diagnostiserats i Sverige. Svårighetsgraden förblir oförändrad genom livet. Om man har en mild form av VWS förblir den mild hela livet. l vissa fall av medelsvår eller mild form kan man se en lindring av sjukdomen med åren, eftersom mängden VWF i blodet kan öka med stigande ålder.
Kan ärftlig form av von Willebrands sjukdom botas?
Nej. VWS kan inte botas i nuläget. Forskning pågår och i framtiden kan det eventuellt finnas sätt att bota sjukdomen med t.ex. genterapi. Det är en livslång, oftast mild rubbning. Dessbättre finns det effektiva och säkra läkemedel för att behandla och förebygga blödningar.
Vilka olika typer av von Willebrands sjukdom finns det?
VWS indelas i tre kategorier typ 1, typ 2 och typ 3. Typ 2 fördelas i sin tur på flera undergrupper (subtyper). När det inte finns tillräckligt med VWF i blodet, eller om VWF inte fungerar normalt, tar det längre tid för blödningar att upphöra.
Vad är typ 1 von Willebrands sjukdom?
Typ 1 är den vanligaste formen av VWS och omfattar ungefär 75 % av alla fall. Vid typ 1 fungerar VWF normalt, men det finns för lite av den i blodet.
Många personer med typ 1 VWS har inga blödningsbesvär alls förrän de råkar ut för en skada eller en operation. Då kan de få en allvarlig blödning.
Andra har milda symtom såsom:
- Blåmärken
- Blödningar från näsa eller mun
- Långvariga blödningar från skärsår.
Vissa kvinnor med VWS har riklig menstruation. Menstruationsblödningarna kan vara så rikliga att de påverkar kvinnans livskvalitet.
Skador eller operationer kan orsaka svåra blödningar även om man har en mild form av VWS. Därför är det viktigt att personer som misstänker att de kan ha VWS blir undersökta.
Vad är typ 2 von Willebrands sjukdom?
Typ 2 VWS är mindre vanlig än typ 1. Ungefär 20–25 % har typ 2.
Vid typ 2 kan mängden VWF i blodet vara normal. Däremot fungerar VWF inte som den ska, vilket ger upphov till ökad blödningsbenägenhet.
Det finns flera undergrupper av typ 2 beroende på vilket slags fel det är på VWF. Det är viktigt att bestämma vilken undergrupp en person med VWS tillhör, eftersom behandlingen kan skilja mellan dem.
Typ 2A är den vanligaste undergruppen och utgör ca 15–20 % av alla fall med VWS. Ungefär 5 % har typ 2B. Personer med typ 2B kan ha brist på blodplättar (trombocytopeni). Typ 2M och 2N är mycket ovanliga undergrupper. Personer med typ 2N, som också kallas ”Normandie”, har som regel normala nivåer VWF, men ovanligt låg nivå av FVIII i blodet. Tillståndet kan därför förväxlas med mild hemofili A.
Vad är typ 3 von Willebrands sjukdom?
Typ 3 VWS är mycket ovanlig och finns endast hos ungefär 1/500 000 individer i befolkningen. Det är den svåraste formen av VWS. Personer med typ 3 har ingen eller nästan ingen VWF i blodet. Därför har de också mycket liten mängd FVIII i blodet, eftersom VWF är ett bärarprotein för FVIII. Dessa personer kan blöda ofta och blödningarna kan bli svåra om de inte behandlas. Spontana ledblödningar och svåra mag- tarmblödningar förekommer. Kvinnor med typ 3 VWS kan behöva behandling med VWF-koncentrat för att dämpa kraftiga menstruationsblödningar.
Översikt von Willebrands sjukdom
|
Typ |
Andel av patienterna |
Svårighetsgrad |
Allmänt |
Blodstillande medel |
|
1 |
75% |
Oftast mild |
Lägre nivå av VWF |
Desmopressin i flertalet fall |
|
2 |
20-25% |
Oftast mild till moderat |
Lägre nivå och defekt funktion av VWF. Flera undertyper |
VWF-koncentrat i flertalet fall. Des mopressin i vissa fall. Desmopressin ska ej användas vid typ 2B |
|
3 |
Enstaka procent |
Svår |
VWF saknas helt och FVIII aktiviteten är mycket låg |
VWF-koncentrat i samtliga fall. Svarar ej på desmopressin |
Läs mer om von Willebrands sjukdom

Ärftlighet
VWS är ärftlig och orsakas av en förändring i arvsanlaget för VWF. Eftersom VWS är ärftlig finns den ofta hos flera medlemmar i samma familj och släkt. Man kan spåra sjukdomen genom att göra ett släktträd.

